I’m playing around with Molly.jl. Not really my field, but some colleagues worked with molecule simulations in the 1990s, so I have been exposed to the basic ideas. I also simulated a set of 5 Argon molecules in 2D, boxed in by a LJ-potential on the wall… (took some 10 minutes with MATLAB in 1994 or so, and some 5 s in Julia a few years back).
So I do the introductory example in the Molly.jl documentation. I have figured how to pick out the resulting vectors from the loggers + construct a time vector for the loggers:
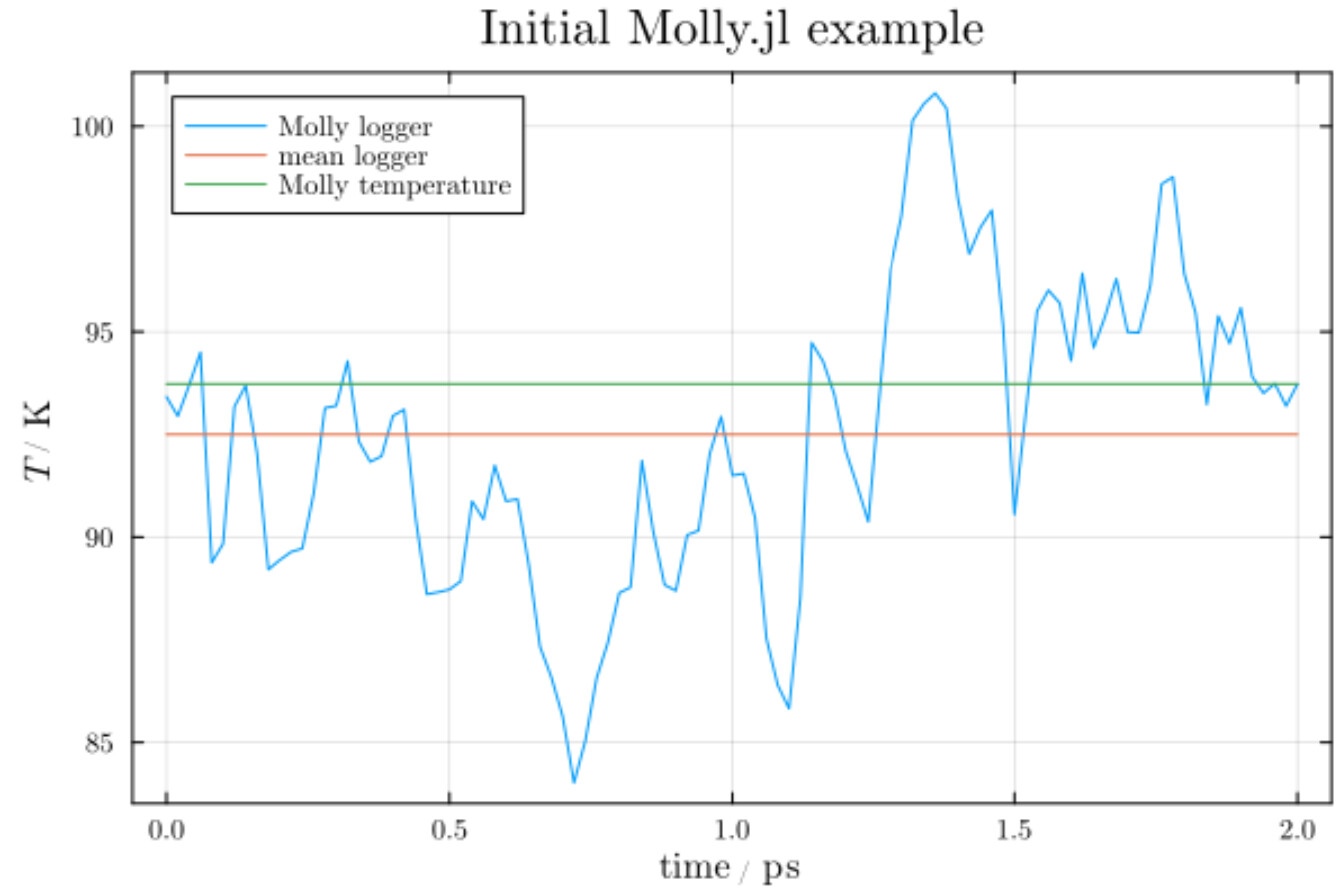
Question 1: I assume the variation in the temperature is a result of the Anderson thermostat not perfectly keeping the temperature at the specified temperature of 100 K?
Question 2: The red line is the mean of the logged temperature evolution. The green line is the result of using Molly’s function temperature(sys).
- I’m curious as to why the green and red lines are different.
Question 3: The temperature is related to the “thermal energy” via the Boltzmann constant, right? Is the variation in temperature a result of variations in particle velocities? Or is it more complex? (Probably…)
I also logged the pressure:

Question 4: Same question as for temperature: is there a simple explanation for why the mean logged pressure and the pressure computed by pressure(sys) are different?
Question 5: The simulation with 100 molecules (atoms) and 1_000 time steps takes some 4 s on my laptop the first time I run the code, and virtually zero upon repeated simulation. Some simple questions:
- how many molecules (and time steps) do I need to get reliable results if I want to compute the compressibility factor z from computed p, T, and molar volume?
- in theory, T should be constant in these simulations, right? the variation is due to imperfect thermostat, right?
- the molar volume is exactly given by the boundary volume and the number of molecules, right?
- if I simulate more complex molecules, I still use keyword “atoms” in the
System“instantiator”, right? - if I have mixtures of molecules, I just include two types of molecules in the “atoms” vector?
Stretching my understanding a bit… If I want to simulate two phase systems (vapor, liquid), do I need to add gravity to get the “liquid” to fall to the bottom? Is it possible to separate density for two (or more) phases?
