Hello yall,
Some coworkers and I have been working on building a quantum chemistry framework in Julia. The package has been registered for a while and I have already had several exchanges with members of the community about it.
Recently, our paper about the program and the general feasibility of Julia to molecular quantum mechanics has been published at the Journal of Chemical Theory and Computation.
I would like to formally introduce Fermi.jl here by highlighting its main features
Basic Infrastructure
-
Molecule files and string (XYZ - Cartesian) format are parsed and processed using the Molecules.jl package, part of the FermiQC environment.
-
Basis set objects and integrals are computed using the GaussianBasis.jl module, also part of the ecosystem. It wraps the C library libcint via libcint_jll, providing a higher-level interface as well as some standard basis set files.
-
Popular ab initio methods
- Restricted Hartree-Fock
- Unrestricted Hartree-Fock
- Moller-Plesset Perturbation Theory (Second Order)
- Coupled Cluster Theory (Up to CCSD(T))
Developent platform
Besides offering basic methods, Fermi.jl aims to serve as a framework for the development of new quantum chemical algorithms. Taking advantage of Julia’s composability, it is possible to add new methods into Fermi without changing it at all (though, if you implement a cool method we will love to incorporate it to the main code!). A guide on how to extend Fermi is available here, using RHF as an example.
Examples
A minimal example of how to compute the electronic energy of water is
using Fermi
@molecule {
O 1.2091536548 1.7664118189 -0.0171613972
H 2.1984800075 1.7977100627 0.0121161719
H 0.9197881882 2.4580185570 0.6297938830
}
@set basis cc-pvtz
@energy ccsd
Macros are just shortcuts to some corresponding function. Sometimes it is advantageous to directly call the functions. For example, the following script plots a potential energy curve for the nitrogen molecule
using Fermi
using Makie
using GLMakie
R = [0.5 + 0.1*i for i = 1:20]
E = Float64[]
@set basis cc-pvdz
for r in R
molstring = """
N 0.0 0.0 0.0
N $r 0.0 0.0"""
Fermi.Options.set("molstring", molstring)
wfn = @energy rhf
push!(E, wfn.energy)
end
f = Figure()
ax = Axis(f[1,1], xlabel="Bond Length (Å)", ylabel="Energy (Hartrees)")
Makie.lines!(R, E, color=:red, linestyle=:dash)
f
which yields

Performance
We performed a series of benchmarks over a set of molecule dimers in the so called S22 data base. For the RHF method, the most basic tool in wave function theory, our algorithm outperforms the one in the well established, C++ written, Psi4 package
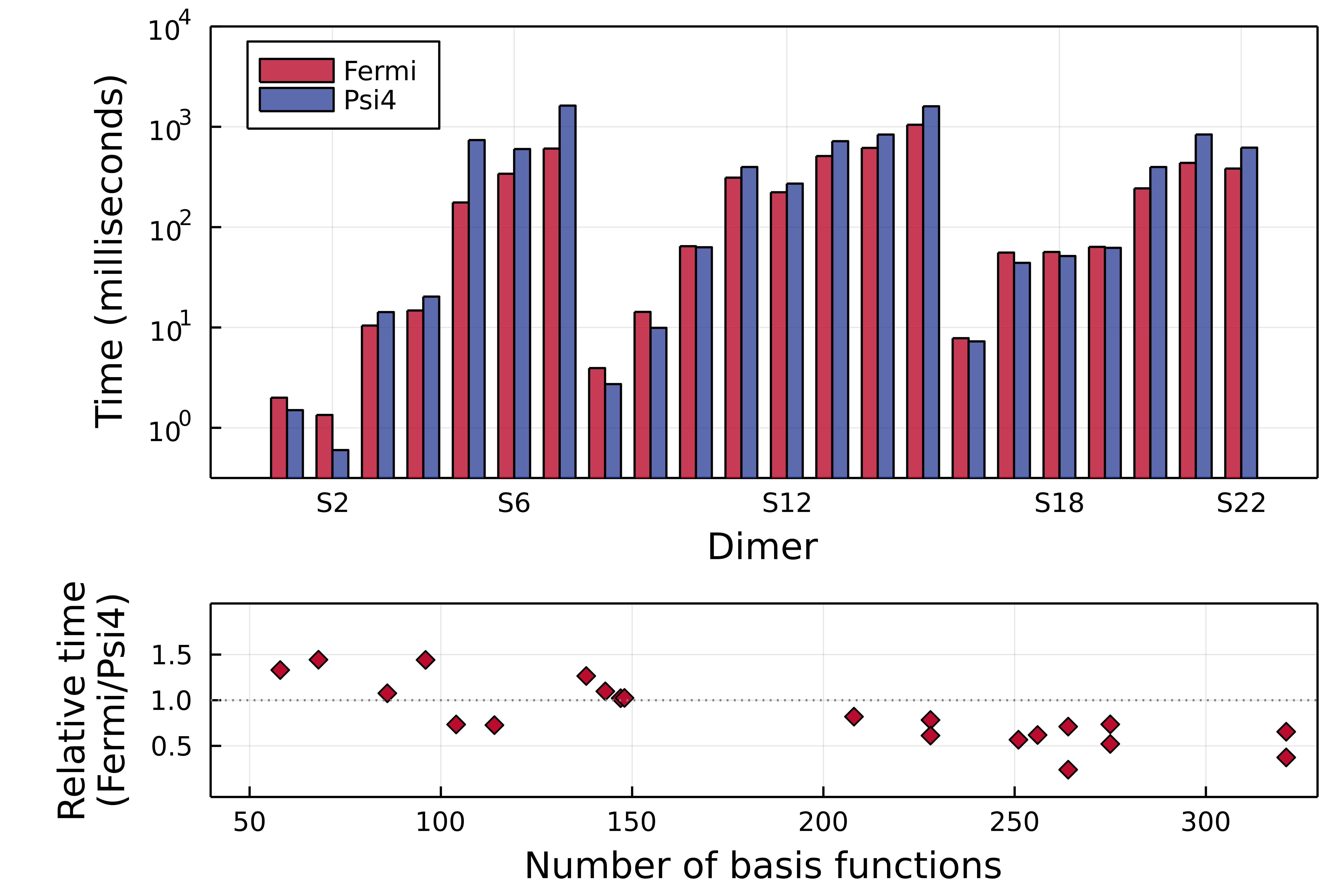
Upper panel shows absolute timings for each iteration of the method over the 22 dimers studied. Lower panel organize these results by the number of basis functions, i.e. the size of the system. The bigger the system, the more demanding the computation is.
During the paper revision, one reviewer wanted to see pure Julia performance since some methods rely on BLAS/TBLIS functions. To offer them a glimpse of what could be done with pure Julia code, I have also used Octavian.jl for matrix multiplications and tensor contraction. I show here one of such results.

Timings to compute the perturbative correction to CCSD - that is, (T). S22 dimers. cc-pVDZ basis set (frozen core).
Future
We are open to suggestions and contributions. The goal is to extend the number of available tools within Fermi and, with time, consolidate its main design. Important features that need to be added include
- Energy gradients
- Unrestricted version of correlated methods
- Wave function analysis tools
- Properties computations
Thank you!
Please reach out if you have any questions!